New publication from the Panasyuk team uncovers a lipid-based defence for damaged lysosomes
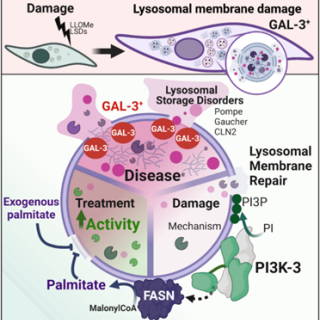
Restoration of lysosomal membrane integrity in cell models of Pompe disease depends on fatty acid synthase and its product palmitic acid
Cellular & Molecular Biology Letters - April 2026
The Panasyuk team reports a new study in Cellular & Molecular Biology Letters revealing how a loss of lysosomal protection may contribute to Pompe disease—and how that protection might be restored.
Lysosomes are the cell’s recycling centres, breaking down unwanted material. In Pompe disease, glycogen accumulates inside lysosomes because it cannot be properly degraded. As this material builds up, lysosomal function deteriorates.
Using cells from patients with Pompe disease, including samples provided by Necker–Enfants Malades Hospital, the team found persistent damage to the lysosomal membrane that became more severe with glycogen accumulation. A key clue was the reduced presence of lipid kinase class 3 PI3K at lysosomes in patient cells. To investigate the mechanism, the team combined a broad range of approaches, including lysosome purification, proteomics and lipidomics. These analyses revealed an unexpected link with lipid metabolism: class 3 PI3K supports fatty acid synthase and the production of palmitic acid, both of which help protect the lysosomal membrane.
Restoring class 3 PI3K directly at lysosomes—or supplying cells with palmitic acid—improved lysosomal degradation and revived cellular recycling. Similar effects in other lysosomal storage disorders suggest that this protective pathway may extend beyond Pompe disease, opening a new translational route towards therapies designed to reinforce damaged lysosomes.
Click here to access the article
This study also reflects a close partnership between clinical activities at Necker–Enfants Malades Hospital and research at INEM. We spoke with first author Edouard Le Guillou, a clinical biologist at Necker and researcher in the Panasyuk team, about the study’s impact and next steps.
Interview – Édouard Le Guillou (INEM)
From clinical metabolism to lysosomal biology: a translational journey
How did you join Ganna Panasyuk’s team?
I first became interested in lysosomal storage diseases during my internship in the Department of Metabolic Biochemistry at Necker–Enfants Malades Hospital, under Professor Jean-François Benoist, where I worked on their diagnosis. At the time, Catherine Caillaud, an MCU-PH in the department, also led the research project on these diseases in Ganna Panasyuk’s lab. Class 3 PI3K, a major focus of Ganna’s team, is a central regulator of cellular organization and lysosomal activity. Disrupting this pathway reproduces several defects seen in lysosomal storage diseases, making it a compelling link between our clinical observations and the lab’s research. Joining the Panasyuk team at INEM for my PhD therefore felt like a natural step, bringing together our clinical expertise, scientific questions and shared interest in lysosomal biology.
What was the most surprising discovery during your PhD?
The turning point came when we analysed proteins from immunopurified lysosomes. The approach was exciting in itself, but the result was completely unexpected: inhibiting class 3 PI3K caused a marked loss of proteins involved in lipid metabolism, particularly fatty acid synthesis. The eureka moment came with the analysis of the purified lysosomes under one specific condition. When we inhibited class 3 PI3K, fatty acid synthase almost disappeared from the lysosomal fraction. It was a striking result that immediately changed the direction of the project. Until then, we had not anticipated such a close connection between lipid metabolism and lysosomal function. That finding opened an entirely new line of investigation and ultimately became one of the central discoveries of the study.
What are the next steps and future perspectives in your research?
I am now a permanent hospital practitioner at Necker–Enfants Malades Hospital, where I diagnose inherited metabolic diseases, while continuing my research in Ganna Panasyuk’s team. This dual role gives me a great opportunity to build a research direction grounded in clinical questions and focused on improving our understanding of lysosomal disorders. Our immediate goal is to test the therapeutic potential of palmitic acid in mouse models of Pompe disease, including its possible use alongside enzyme replacement therapy. More broadly, I want to uncover further secrets of the lysosome—not simply as a recycling compartment, but as a remarkable metabolic hub where cells sense nutrients, break down macromolecules and make key metabolic decisions. I also aim to develop new diagnostic and follow-up biomarkers, including galectin-3 and candidates emerging from lysosomal multi-omics. I am genuinely excited to continue growing as both a clinician and a scientist, to shape this research programme within the Panasyuk team, and to contribute to ambitious, collaborative science across our campus.